Research
Recent researches:
The object of my research is to understand molecular-level dynamics of polymer systems, including associative polymers, polymer blends and polymer nanocomposites. My research interests focus on several areas below:
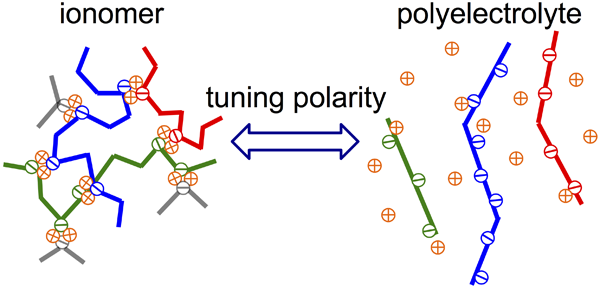
Determination and tuning the interaction energy of associative polymers: My research works have contributed to understanding how the interactions among polymer chains influence statics, dynamics, and application of the materials. My recent focus is placed on associative polymers containing ionic or hydrogen bonding groups as stickers, for which the most important parameter is the interaction energy between associative groups.[1-3] To determine the energy is a long-term challenge in the field of associative polymers. Using rheology, we characterized stress relaxation originating from both the thermal motion and sticker dissociation. We found that the interaction energy can be consistently determined from a delay of the sticker dissociation with respect to the thermal motion of Kuhn segment, as well as a ratio of temperature dependences of these two types of motion. This protocol is superior to the conventional method that relies on the temperature dependence of viscosity, which tends to overestimate the interaction energy. Considering the electrostatic nature of ionic interaction, we designed a model system enables precise tuning of polarity of the medium, and accordingly the ion interaction energy.[4, 5] More importantly, the study of this system revealed a transition region from ionomer to polyelectrolyte (see Figure below), a previously no man’s land in the studies of ion-containing polymers.
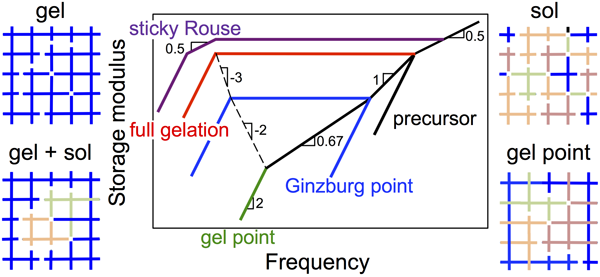
Gelation of associative polymers: It is well known that strongly associative polymers form physical sol or gel. Then, a very natural and fundamental question is, where is the sol-to-gel transition point? My research works have greatly advanced the understanding of this transition.[3, 6, 7] In collaboration with Dr. Weiss in Akron University and Dr. Colby in the Pennsylvania State University, we precisely synthesized ionomers with an average number of stickers per chain from less than one to more than two. From their linear viscoelasticity we observed, to the first time, dynamic change of the ionomers along with a sol-to-gel transition. This transition can be rationalized if we consider each monomer of the ionomer as a functionizable group (through ionization). We also modified the mean-field theory of Rubinstein and Semenov by introducing the Ginzburg transition.[3] The newly developed reversible gelation model can predict well the linear viscoelasticity observed in experiments. Recently, we extended this approach to varied associative systems, including multiple hydrogen bonding system[1] and associative polymer solutions with randomly positioned stickers.
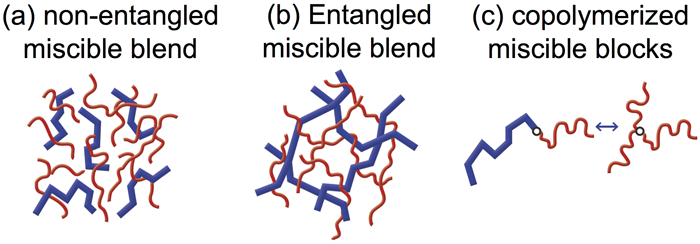
Relationship between mechanical reinforcement and ionic transportation: For ion-containing polymers, the strengthening of ionic interaction can reinforce the materials. In contrast, the weakening of ionic interaction can enable the ions to dissociate and transport within the polymer medium.[8-10] The resulting ion-conductive materials are widely used in ion battery, fuel cell, supercapacitors, sensors and actuators. The stress reinforcement and ion conduction are a long-term contradiction in the battery industry; improving one of them usually sacrifices the other. For a series of poly(ethylene oxide) based ionomers, I found that the ionic fluctuation time, detected in the dielectric spectroscopy, coincided with the relaxation time of sticky Rouse segment detected in the linear viscoelasticity. This finding explained, to the first time, the well-known contradiction from a molecular level. Namely, the hopping of ionic groups from one cluster to another is coupled with the motion of sticky Rouse segments. With this knowledge, we realized a prediction of the linear viscoelasticity from the dielectric spectroscopy. This work has been collaborated with Dr Colby and highlighted in his 2013 Bingham Medal award speech.[9] Dynamic asymmetry of polymer blends and copolymers: Under the guidance of Dr Watanabe in Kyoto University, I improved the understanding of dynamic asymmetry in miscible polymer pairs.[11-14] We designed a model system where the fast component have dipoles parallel along the chain backbone, whose end-to-end fluctuation can be selectively detected in the dielectric spectroscopy. A combination of the dielectric and rheological measurements enables a separation of the dynamic behavior of the two components. Studies of this model system revealed three important dynamic features: (a) the composition fluctuation of slow component can be quenched to a scale as large as the chain dimension,[11] (b) a localized motional coupling of two components significantly modifies the relaxation dynamics below the entanglement length, where the fast component is forced to equilibrate together with the slow component.[13] (c) when the two dynamic asymmetric blocks are combined in a block copolymer, the slow block would anchor the motion of the fast block, to force the latter to relax like a star arm. This result evidences that “free linear chain” can also exhibit tethered feature.[12] These works have been highlighted in Dr Watanabe’s 2016 Bingham Medal award speech.
1. Z. J. Zhang, C. Liu, X. Cao, L. C. Gao and Q. Chen, Macromolecules, 2016, 49, 9192-9202.
2. Z. Zhang, C. Huang, R. A. Weiss and Q. Chen, J Rheol, 2017, 61, 1199-1207.
3. Q. Chen, C. W. Huang, R. A. Weiss and R. H. Colby, Macromolecules, 2015, 48, 1221-1230.
4. Q. Chen, N. Bao, J.-H. H. Wang, T. Tunic, S. Liang and R. H. Colby, Macromolecules, 2015, 48, 8240-8252.
5. Z. Zhang, C. Liu, X. Cao, J.-H. H. Wang, Q. Chen and R. H. Colby, Macromolecules, 2017, 50, 963-970.
6. C. W. Huang, Q. Chen and R. A. Weiss, Macromolecules, 2016, 49, 9203-9214.
7. C. W. Huang, C. Wang, Q. Chen, R. H. Colby and R. A. Weiss, Macromolecules, 2016, 49, 3936-3947.
8. Q. Chen, S. Liang, H.-S. Shiau and R. H. Colby, ACS Macro Letters, 2013, 2, 970-974.
9. Q. Chen, G. J. Tudryn and R. H. Colby, J Rheol, 2013, 57, 1441-1462.
10. Q. Chen, H. Masser, H.-S. Shiau, S. Liang, J. Runt, P. C. Painter and R. H. Colby, Macromolecules, 2014, 47, 3635-3644.
11. Q. Chen, Y. Matsumiya, Y. Masubuchi, H. Watanabe and T. Inoue, Macromolecules, 2008, 41, 8694-8711.
12. Q. Chen, Y. Matsumiya, Y. Masubuchi, H. Watanabe and T. Inoue, Macromolecules, 2011, 44, 1585-1602.
13. H. Watanabe, Q. Chen, Y. Kawasaki, Y. Matsumiya, T. Inoue and O. Urakawa, Macromolecules, 2011, 44, 1570-1584.
14. Q. Chen, Y. Matsumiya and H. Watanabe, Polym J, 2012, 44, 102-114.